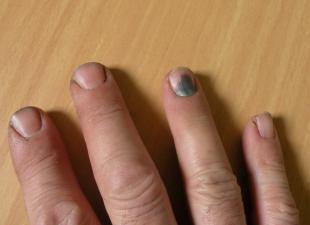
تريد كل أم أن ينمو طفلها بصحة جيدة ويتطور حسب عمره.
ولهذا السبب يولي الآباء دائمًا اهتمامًا خاصًا ارتفاع الطفل لأن الطول هو أحد المؤشرات الرئيسية لنمو الطفل.
كيف يجب أن ينمو الطفل من عمر 3 إلى 7 سنوات؟ ماذا تفعل إذا أن يكون الطفل قصيرًا جدًا أو على العكس طويلًا جدًا ؟ ما الذي يمكن أن يسبب اضطرابات النمو؟
ندعوك لتكتشف معنا اليوم إجابات هذه الأسئلة المهمة.
معايير النمو
أسباب اضطرابات النمو
من بين الأسباب الرئيسية لاضطرابات النمو ما يلي:
- - إذا كانت الأم والأب قصيرين أو طويلين على التوالي، فيمكن للطفل ببساطة أن يحذو حذو والديه ويكون قصيرًا أو طويلًا؛
إيرينا كولباكوفا، طبيبة أطفال، معالجة تجانسية - مركز المعالجة المثلية الذي سمي على اسمه. ديميانا بوبوفا: "إذا كانت أم الطفل أو أبيه قصير القامة، فمن الواضح أنه لن يكبر طويل القامة، فلا حرج في ذلك. من بين الأشخاص الناجحين المشهورين هناك العديد من الأشخاص القصيرين ولكن ليس أقل جمالا وطموحا. على سبيل المثال، نابليون، توم كروز، شاكيرا، فانيسا باراديس، سلمى هيك، بابلو بيكاسو، مارغريت ميتشل، شارلوت برونتي."
- مشاكل هرمونية - يمكن أن تؤدي إلى نمو مرتفع جدًا أو قصير جدًا، على سبيل المثال، يمكن أن تسبب العقدة والتقزم؛
- الأمراض والاضطرابات الخلقية - على سبيل المثال، الودانة هي مرض خلقي لا يكون لدى الشخص مناطق نمو في عظام الأطراف، مما يؤدي إلى نمو طول الذراعين والساقين؛ متلازمة مارفان هي مرض وراثي يصيب النسيج الضام، ويتميز المرضى بقامة طويلة وأطراف ممدودة.
- اضطرابات النمو - تأخر النمو أو، على العكس من ذلك، التطور السريع للغاية يمكن أن يسبب نموا منخفضا أو مرتفعا؛
- سوء التغذية والأمراض المزمنة والإجهاد الشديد يمكن أن تؤثر أيضًا على نمو الطفل.

نمو الطفل: نصيحة للآباء
في معظم الحالات طول الطفل قصير أو طويل في عمر أو آخر، تكون هذه سمة من سمات الطفل ولا يعاني من أي مشاكل صحية أساسية. ولكن في بعض الأحيان تكون أمراض مختلفة هي المسؤولة عن اضطرابات النمو.
لفهم ما إذا كان نموه صحيحا أم طبيعيا، فمن الضروري استشارة طبيب الأطفال.
يجب على الطبيب تقييم الحالة العامة للطفل، ومراقبة ديناميكيات نموه لعدة أشهر، وتقييم العوامل الوراثية والعيوب الخلقية المحتملة، ثم التوصل إلى استنتاج حول ما إذا كان الطفل يتطور بشكل صحيح. إذا كانت هناك انحرافات حقيقية، فسيقوم الطبيب بإحالة الطفل للفحص إلى طبيب الغدد الصماء وغيره من المتخصصين المتخصصين سوف يصف العلاج المعقد .
ولمنع مشاكل النمو، يُنصح الآباء بما يلي:
- يزور طبيب الأطفال 5 مرات على الأقل في السنة للفحص الروتيني؛
- أن تكون مهتمًا بمعايير النمو؛
- توفير للطفل التغذية السليمة وأنماط المشي والنوم المثالية ;
- مهم جدا النشاط البدني المعتدل ;
- إذا كان ذلك ممكنا فمن الضروري حماية طفلك من التوتر وخاصة قبل سن الثالثة.
إذا كانت لديك شكوك حول ما إذا كان طفلك يتطور بشكل صحيح، فتأكد من استشارة الطبيب لفهم الوضع وعدم تفويت الوقت الذي يمكن فيه تصحيح الأمراض الخطيرة.
قبل أن تشعري بالذعر بشأن نمو طفلك، ألقِي نظرة هادئة على الوضع واستشيري الطبيب واستخلصي النتائج. لا داعي للقلق، فطفلك يحتاج حقًا إلى أم هادئة وصحية. تذكر أن كونك قصير القامة وطويل القامة لهما إيجابيات وسلبيات!
كن بصحة جيدة!
يمكن أن يحدث الطول لأسباب مختلفة - وراثية وغير وراثية.
الأسباب الوراثية لطول القامة
متلازمة كلاينفلترغالبًا ما يحدث بسبب النمط النووي XXY، ولكن قد يحدث النمط النووي XXYY وأشكال الفسيفساء. (مع النمط النووي XXXY، من المرجح أن يكون النمو بطيئًا.) هناك ميل نحو التخلف العقلي الخفيف، والذي غالبًا ما يتفاقم بسبب الاضطرابات السلوكية، وقصور الغدد التناسلية المفرط. في الأعمار الأكبر، هناك احتمال كبير للإصابة بمرض السكري. عادة ما يتم ملاحظتها، ولكن تم وصف حالات نجاح حقن الحيوانات المنوية داخل الهيولى.
الأولاد الذين لديهم النمط النووي XYY لديهم تأخر معتدل في النمو الفكري ومشاكل محددة في التنسيق الحركي. في الماضي، كان يُعتقد أن هؤلاء المرضى هم أكثر عرضة لإظهار السلوك المعادي للمجتمع، ولكن هذا الأمر أصبح موضع تساؤل الآن. قد يحدث الخصية الخفية، لكن هذا العرض ليس شائعًا كما هو الحال في متلازمة كلاينفلتر.
كلا الحالتين شائعتان نسبيًا، تقريبًا ضعف شيوع متلازمة تيرنر (استنادًا إلى الدراسات السكانية للنمط النووي عند الولادة)، لكن صعوبات التعلم البسيطة والإدراك العام الطبيعي للطول يؤديان إلى اكتشاف المرض في أواخر مرحلة الطفولة أو مرحلة البلوغ المبكر.
المظاهر المظهرية الخارجية لدى النساء ذوات النمط النووي XXX قليلة: هناك ميل نحو نقص الوزن وطول القامة، وفي بعض المرضى يحدث البلوغ المتأخر وانقطاع الطمث والعقم. متوسط معدل الذكاء (حاصل الذكاء) هو 85 نقطة.
متلازمات التشوه الناجمة عن اضطرابات التمثيل الغذائي أو الأنسجة الضامة
متلازمة مارفان هي اضطراب وراثي شائع نسبيًا حيث يتم تغيير نسخة واحدة من جين الفيبريلين الموجود على الكروموسوم 17q. يتميز المرضى بقامة طويلة غير متناسبة مع أرجل طويلة نسبيًا، وعنكبوتية الأصابع، وفرط حركة المفاصل، والفتق، والجنف وتشوهات الصدر، وقصر النظر، وخلع أو خلع جزئي للعدسة، والحنك القوطي. قد تكون المظاهر من الأعضاء الداخلية عبارة عن ضعف في هياكل الكولاجين،
خاصة في الجانب الأيسر من القلب، ويتجلى ذلك في قصور الصمام التاجي والأبهري، وتوسع، وتسلخ الأبهر. قد تحدث عفوية أيضا. يكشف التصوير بالرنين المغناطيسي عن نتوءات الأم الجافية في المنطقة القطنية العجزية.
تقلص بيل العنكبوتي هو اضطراب وراثي نادر يشترك في بعض الميزات مع متلازمة مارفان وينتج عن خلل في نسخة أخرى من جين الفيبريلين الموجود على الكروموسوم 15q. تسبب هذه الحالة تقلصات في الركبتين والمرفقين واليدين، وصغر الفك، وشكل غير طبيعي لأذرع الأذن ("الأذنين المسطحة") والجنف الحدابي.
بيلة هوموسيستينية هو مرض من مجموعة أمينوسيدوريا، حيث يلاحظ نوع طويل القامة يشبه مارفان. يتم تشخيص هذا المرض في كثير من الأحيان أكثر من متلازمة مارفان بسبب مضاعفات العين مثل عدسة خارج الرحم وقصر النظر الشديد. عادة ما يتم تقليل الذكاء، وتحدث مضاعفات بسبب الجلطات الدموية.
يتميز الحثل الشحمي المعمم بالتخلف الواضح في الدهون تحت الجلد والنمو المرتفع نسبيًا.
متلازمات التشوه مع نوع متماثل من القامة الطويلة
معظم هذه المتلازمات (المذكورة أدناه) تقترن بتأخر النمو الفكري.
متلازمة سوتوس.
متلازمة ويفر.
متلازمة مارشال سميث.
متلازمة بيكويث-فيدمان، التي تتميز بالعملقة (في كثير من الأحيان أكثر من ذلك).
تظهر على جانب واحد)، وعلامات تشوه أخرى ونقص السكر في الدم. قد تكون نتيجة نقص السكر في الدم ضعفًا فكريًا. هناك أدلة لصالح زيادة التعبير عن عامل النمو IGF-2 في فترة ما قبل الولادة، والتي قد تترافق مع ميل لتطوير ورم ويلمز.
متلازمة سيمبسون-جولابي-بيميل - "متلازمة البلدغ" لها نفس المظاهر جزئيًا. يعتمد المرض على تنشيط الطفرات في الجين glypican 3، وهو بروتين غليكان مرتبط بالغشاء ومسؤول عادة عن عزل IGF-2 والحد من توافر الأخير لمستقبله.
يمكن ملاحظة الطول أيضًا في متلازمة الكروموسوم الهش، ومتلازمة بانايان-رايلي-روفالكالبا، ومتلازمة إليالد، ومتلازمة نيفو.
متلازمات التشوه مع تضخم جزئي أو غير متماثل لأجزاء الجسم
يمكن ملاحظة زيادة في أجزاء الجسم من نوع الضخامة النصفية في متلازمة بيكويث-فيدمان. تتميز متلازمة كليبل-ترينوناي-ويبر بمزيج من أجزاء الجسم المتضخمة مع الشامات الوعائية الجلدية. يعتمد هذا العلاج على خلل في الأنسجة الخيمرية، مما يؤدي إلى نمو تدريجي وأورام شحمية وتشوهات.
الأسباب الثانوية لطول القامة
فرط الانسولينية
نظرًا لأن الأنسولين يعمل كعامل نمو، فإن فرط أنسولين الدم داخل الرحم، والذي يتطور استجابةً لمرض السكري لدى الأم، أو نقص السكر في الدم الناتج عن فرط أنسولين الدم المستمر عند الوليد (PHHN)، والذي كان يُسمى سابقًا خلل تنظيم الغدد الصماء البنكرياسية أو داء الأرومة غير المتجانسة، يؤدي إلى تطور العملقة. عادة ما تكون الزيادة في الحجم عابرة، وبمجرد القضاء على مصدر إفراز الأنسولين المرضي، يلاحظ تباطؤ في النمو.
قد يحدث فرط الأنسولينية بشكل ثانوي للسمنة. يساهم الإفراط في تناول السعرات الحرارية في نمو الطفل وقد يعجل بفرط الأنسولينية، مما يسبب الطول النسبي، الذي يتميز بالطول عند الطرف العلوي من النطاق المستهدف الطبيعي، والوزن المئوي الذي يتجاوز الطول المئوي، والبلوغ المبكر نسبيًا، وعلامات التمدد، والاحمرار اللامع الذي يشبه متلازمة كوشينغ الخفيفة (ومع ذلك، كل هذا على خلفية النمو المتسارع، الذي يميز هذه الحالة عن فائض هرمونات الستيرويد، حيث يوجد دائمًا تباطؤ في النمو). غالبًا ما يكون هناك تشابه خارجي بين أحد الوالدين والأشقاء أو كليهما. في حالة فرط الأنسولينية الشديدة، يمكن أيضًا ملاحظة الشواك، وفي متلازمة HAIR-AN؛ توصف مظاهر الوجه بأنها "ضخامة النهايات". الأورام والخلل الوظيفي في منطقة ما تحت المهاد يمكن أن يسبب الإفراط في تناول الطعام والسمنة وزيادة الطول كظاهرة ثانوية.
الانسمام الدرقي
يؤدي التسمم الدرقي الخفيف، ونتيجة لذلك، غير المعترف به وغير المعالج، إلى النمو المتسارع والطول النسبي في مرحلة الطفولة المتوسطة. ومع ذلك، في هذه الحالة، يكون هناك تقدم في عمر العظام، لذا فإن الارتفاع النهائي، كقاعدة عامة، يقع ضمن النطاق الجيني.
البلوغ المبكر
ويتميز هذا المرض بالنمو المرتفع في مرحلة الطفولة، ولكن ليس في مرحلة البلوغ. إذا لم يتم علاجه، يؤدي النضج العظمي المتقدم إلى الإغلاق المبكر لصفائح النمو المشاشية، والتي تظهر عادة على شكل قصر القامة في مرحلة البلوغ.
الحالات الأخرى المرتبطة بحجم الجسم غير المتناسب والطول النسبي
الورم الغدي الغدد الصماء المتعدد (MEA أو MEN) من النوع IIb هو مرض عائلي يقترن فيه الورم الغدي النخاعي بقامة متوسطة وملامح تشبه مارفان، بالإضافة إلى أورام عصبية في الأغشية المخاطية والأمعاء والملتحمة.
يمكن أن يسبب قصور الغدد التناسلية زيادة معتدلة في الطول النهائي مع زيادة نسبية في طول الساق (ما يسمى بالجسم المخصي)، والذي يحدث بسبب الإغلاق المتأخر للمشاشات والنمو المطول للأطراف في مرحلة الطفولة، بالإضافة إلى التأثير غير الكافي للجنس الهرمونات على نمو العمود الفقري.
وبالمثل، ولكن بدرجة أكثر شدة، يتداخل نقص الأروماتاز مع تحويل هرمون التستوستيرون إلى هرمون الاستروجين لدى الرجال (كما هو الحال مع عيوب مستقبلات هرمون الاستروجين)، وبالتالي عدم تحفيز إغلاق لوحة نمو المشاشية. في هذه الأمراض النادرة، يستمر المرضى في النمو حتى مرحلة البلوغ؛ لديهم هشاشة العظام والعقم، بالإضافة إلى زيادة كبيرة في تركيز الهرمون المنبه للجريب (FSH).
قد تترافق المقاومة العائلية للجلوكوكورتيكويدات مع القامة الطويلة.
خوارزمية لتشخيص القامة الطويلة
التاريخ والفحص البدني
عند جمع سوابق المريض من طفل تظهر عليه علامات القامة الطويلة، ينبغي توضيح النقاط الأساسية التالية.
حجم الجسم عند الولادة، صحة الأم أثناء الحمل، مسار الحمل، نوع الولادة. هل كانت الأم رجولة أثناء الحمل؟ (= نقص الأروماتيز عند الطفل).
حجم الجسم وتوقيت البلوغ الأبوي.
أمراض القلب المبكرة وأمراض العين لدى أي فرد من أفراد الأسرة. عائلة
تاريخ أورام الغدد الصماء وأمراض الغدد الكظرية (MEN-IIb، المقاومة العائلية للجلوكوكورتيكويدات).
أي أعراض تشير إلى البلوغ المبكر.
أي من الأعراض التالية: التعرق الزائد، الرعشة، حركات الأمعاء المتكررة، القلق، أو عدم تحمل الحرارة.
استهلاك الطعام.
أعراض عصبية، بما في ذلك الاضطرابات البصرية. هل حاسة الشم لديك محفوظة؟ (يرتبط فقدان حاسة الشم بقصور الغدد التناسلية في متلازمة كالمان).
مستوى التطور أو التعليم. هل هناك أي اضطرابات حركية أو سلوكية معينة؟
عند إجراء فحص موضوعي للمكانة العالية، يتم إيلاء اهتمام خاص للمؤشرات التالية.
ديناميات نمو الوقوف والجلوس ووزن الجسم ومحيط الرأس. يعد وجود أو عدم التناسب علامة مهمة. في متلازمة سوتوس، يزداد محيط الرأس، ولأن الانحراف المعياري للطول (SDS) لم يعد يتغير بعد سن الثانية، لم يعد هؤلاء الأطفال يبدون طويلي القامة.
غالبًا ما يتم ملاحظة السطور الأفقية على جلد الظهر عند الأطفال ذوي النمو السريع، بغض النظر عن سببها.
ميزات التشوه.
الأورام العصبية أو تضخم الغدة الدرقية أو ارتفاع ضغط الدم (الانتيابية في المراحل المبكرة) في MEN-IIb.
أي علامات على قصور الغدد التناسلية أو الخصية الخفية. الخصية الكبيرة (لوحظ في متلازمة X الهشة، نقص الأروماتيز).
ويلاحظ وجود آذان كثيرة ومشعرة عند الرضع الذين يولدون لأمهات مصابات بداء السكري.
تلطيخ النخيل (مع العملقة).
محدودية المجال البصري، نوع العصب البصري، موضع العدسة.
تضخم الغدة الدرقية، ورعاش، وجحوظ أو غيرها من علامات التسمم الدرقي.
تفسير البيانات التي تم الحصول عليها
عدم وجود أي تشوهات أو اختلالات جسدية في حالة عدم وجود علامات البلوغ المبكر: إذا كان وزن الجسم مئوياً< роста = семейная высокорослость; если перцентили массы тела >الطول (أو القيمة تتجاوز 97% على منحنى وزن الجسم مقابل الطول) = علم الأمراض الغذائي.
حجم الجسم الكبير مصحوبًا بأعراض تشوه محددة وضعف فكري: إحدى المتلازمات المرتبطة بالقامة الطويلة.
بنية غير متناسبة مع ذكاء طبيعي: وجود العنكبوتية = متلازمات مارفان أو بيلز؛ عند حدوث طفرة جديدة، قد لا تكون هناك حالات للمرض في تاريخ العائلة؛ مكانة طويلة معتدلة، ورم عصبي في الشفة أو اللسان أو الجفن، وتاريخ عائلي (على الرغم من شيوع طفرات جديدة) = MEN IIb؛ القامة المعتدلة، قصور الغدد التناسلية وفقدان حاسة الشم = متلازمة كالمان؛
القامة المعتدلة وقصور الغدد التناسلية = قصور الغدد التناسلية المعزول أو علاجي المنشأ (على سبيل المثال، بسبب الإشعاع)؛ نقص الاروماتيز.
بنية الجسم غير المتناسبة مع التخلف العقلي: قصور الغدد التناسلية أو الخصية الخفية = مضاعفة الكروموسوم X؛ إذا ظهرت أمراض العيون والجهاز العصبي في المقدمة = بيلة هوموسيستينية. متلازمة X الهشة. تضخم جانب واحد من الجسم أو أحد الأطراف: عندما يقترن بعلامات التشوه الواردة في الجدول. 3.1، = متلازمة بيكويث-فيدمان؛ عندما يقترن بمتلازمة كليبل-ترينوناي-ويبر؛ عند دمجها مع الشامات الخطية والأورام الشحمية = متلازمة بروتيوس.
حجم الجسم الكبير في حالة عدم التناسب: إذا ظهر منذ الولادة = فرط الأنسولينية الوليدية؛ مع زيادة معدل النمو، والأعراض العصبية لضغط التصالب البصري أو أعراض الجلد = عملاقة الغدة النخامية. في وجود تضخم الغدة الدرقية، جحوظ، رعاش، عدم انتظام دقات القلب = الانسمام الدرقي. مع البلوغ المبكر (قبل 8 سنوات للفتيات و 9 سنوات للصبيان) = البلوغ المبكر.
الأشعة السينية والفحوصات المخبرية
تكون الأشعة السينية والفحوصات المخبرية مطلوبة بشكل أقل في حالات القامة الطويلة مقارنة بحالات قصر القامة.
يمكن للأشعة السينية لعظام اليد والمعصم لتحديد عمر العظام أن تساعد في تقييم النضج الفسيولوجي وتأكيد العنكبوتية. لتحديد نتوءات الأم الجافية في المنطقة القطنية العجزية في متلازمة مارفان، يلزم إجراء تصوير بالرنين المغناطيسي، لكن مؤشرات هذه الدراسة في غياب الأعراض العصبية في الأطراف السفلية نادرة.
يتسارع عمر العظام بشكل معتدل مع القامة العائلية الطويلة (البلوغ المبكر) وبشكل أكثر أهمية مع البلوغ المبكر والتسمم الدرقي.
لوحظ تسارع واضح في عمر العظام في متلازمة مارشال سميث. بينما في متلازمات التشوه الأخرى المصحوبة بتضخم الجسم، يتسارع عمر العظام بدرجة أقل. في متلازمة ويفر، يتم تسريع نضوج عظام الرسغ بالنسبة إلى عظام اليد الصغيرة.
يتم حساب مؤشر المشط كمتوسط نسبة طول وعرض عظام المشط II و V. يتم تعريف العنكبوتية عندما تكون قيمة المؤشر أعلى من 8.5، على الرغم من أن هذا في الواقع يضيف القليل إلى نتائج التقييم السريري بناءً على علامات خارجية.
يعد وجود الأعضاء التناسلية المتغيرة بشكل مرضي مؤشراً على التنميط النووي.
إذا تم الاشتباه في MEN-IIb بناءً على تاريخ عائلي للمرض أو وجود أورام عصبية على الأغشية المخاطية لدى طفل ذو نمط ظاهري يشبه مارفان، فمن الضروري تحديد تركيز الكالسيتونين في الدم وحمض الفانيليل مانديليك بسرعة في الدم. البول وتأكيد التشخيص عن طريق تحليل الجين الورمي الأولي (RET) على الكروموسوم 10q، لأن التشخيص المتأخر لسرطان الغدة الدرقية النخاعي يمكن أن يكون له عواقب وخيمة.
يمكن تأكيد فرط الأنسولينية الوليدية لدى الأطفال المولودين لأمهات غير مصابات بداء السكري عن طريق تحديد مستويات الأنسولين المرتفعة بشكل غير مناسب لنقص السكر في الدم.
إذا كان هناك شك، تتم الإشارة إلى اختبار وظائف الغدة الدرقية للتأكد من وجود مستويات TSH المكبوتة؛ الأمر نفسه ينطبق على اختبار البول لتحديد مستويات الهوموسيستين.
في حالة الاشتباه في عملاقة الغدة النخامية، فقد تكون مستويات IGF-1 المرتفعة بمثابة اختبار فحص مفيد. بالإضافة إلى ذلك، يتم استخدام تحديد ملف الإفراز الفسيولوجي لهرمون النمو (هرمون النمو)، ويتم أيضًا تأكيد عدم وجود قمع لهرمون النمو عن طريق حمل الجلوكوز.
تم الكشف عن التنافر أحادي الوالدين (الذي يسبب تعطيل بصمة IGF-2 على الذراع القصير للكروموسوم 11) في 80٪ من حالات متلازمة بيكويث-فيدمان.
علاج
إن علاج طول القامة لدى الأطفال بهدف تقليل الطول النهائي هو علاج متخصص للغاية ويجب إجراؤه فقط في المراكز ذات الخبرة في هذا العلاج.
مع القامة الطويلة مجهولة السبب، يمكن تعريف الطول النهائي الذي يزيد عن 185 سم عند النساء و200 سم عند الرجال بشكل مشروط على أنه "مفرط"، على الرغم من أن الكثير يعتمد على التكيف النفسي للطفل والدعم من الوالدين والأقران. سيكون من الخطأ معاملة الطفل بسبب تصور غير مناسب للطول أو تجارب سابقة سلبية لأحد الوالدين.
إن تحفيز البلوغ المتسارع بشكل مصطنع قد يقلل من الطول النهائي إلى حد ما. يتم تحقيق ذلك عادة عن طريق وصف جرعات يومية عالية من إيثينيل استراديول (10-200 ملغ عن طريق الفم) للفتيات ومستودعات هرمون التستوستيرون عن طريق الحقن للأولاد (تصل إلى 500 ملغ كل أسبوعين). ويرتبط هذا النظام العلاجي للطول بالمشاكل النفسية المرتبطة بالبلوغ المفاجئ، بالإضافة إلى الآثار الجانبية مثل الألم في الغدد الثديية والأعضاء التناسلية، والظهور المفاجئ لحب الشباب، وما إلى ذلك. مجموعة التستوستيرون، يوصف تطور القساح. في الفتيات اللاتي يتلقين علاج الاستروجين، الجلطات الدموية. لا تزال مخاطر الإصابة بآثار جانبية طويلة المدى غير معروفة وتسبب القلق، خاصة عند الفتيات اللاتي لديهن تاريخ عائلي للإصابة.
تجري حاليًا تجارب سريرية باستخدام نظير السوماتوستاتين طويل المفعول المؤتلف، والذي يمثل نهجًا فسيولوجيًا أكثر لعلاج القامة الطويلة، لكن النتائج طويلة المدى لهذا العلاج لا تزال غير واضحة.
يُقترح في كثير من الأحيان أن العلاج بالهرمونات الجنسية لدى المرضى الذين يعانون من متلازمة مارفان يجب أن يتم بحذر بسبب الزيادة الموجودة نظريًا في خطر الإصابة بأمراض القلب والأوعية الدموية، على الرغم من عدم وجود دليل منشور لهذا الموقف.
عندما يتم تشخيص القامة الطويلة ومرض MEN IIb، تتم الإشارة إلى استئصال الغدة الدرقية الوقائي في حالات الطوارئ، يليه علاج بديل باستخدام ليفوثيروكسين الصوديوم وفيتامين د ومكملات الكالسيوم، بالإضافة إلى المراقبة مدى الحياة للكشف المبكر عن ورم القواتم.
في معظم الأولاد ذوي القامة الطويلة وازدواج الكروموسوم X، يشار إلى العلاج ببدائل التستوستيرون لتحقيق تطوير الخصائص الجنسية الثانوية وتقليل عدم التناسب. يتم تحقيق ذلك عادةً عن طريق إعطاء هرمون التستوستيرون (50-100-250 مجم بالتتابع)، على الرغم من أنه يمكن استخدام اللاصقات والأدوية عن طريق الفم في بعض المراكز. كل هذه العلاجات قد تؤدي إلى تفاقم المشاكل السلوكية المرتبطة بطول القامة.
في المتلازمات المختلفة المصحوبة بقامة طويلة، تمت محاولة الاستئصال الجراحي لأجزاء من العظام الأنبوبية الطويلة لتقليل الارتفاع النهائي، والتي، مع ذلك، حققت نجاحًا محدودًا وأدت إلى إعاقة متكررة في القامة الطويلة بسبب تطور عدم التماثل. قد يؤدي حفر المشاش في الظنبوب العلوي وعظم الفخذ السفلي إلى نتائج أكثر إيجابية مع الخبرة المناسبة في مثل هذه التدخلات.
تم إعداد المقال وتحريره بواسطة: الجراحمميزات تشخيص وعلاج طول القامة عند الأطفال
ووفقا للدراسات الوبائية، فإن معدلات النمو المرتفعة شائعة لدى الأطفال ونادرا ما تكون سببا لطلب الرعاية الطبية. عادة، يطلب الآباء المشورة من طبيب الغدد الصماء لدى الأطفال بشأن القامة الطويلة بسبب وجود الأعراض المصاحبة: التعب، والإغماء، واضطرابات الوضع المختلفة، وتأخر أو تسارع النمو الجنسي. ومع ذلك، في المراهقين ذوي القامة الطويلة، غالبا ما يكون هناك تأخر في تكوين نظام الأوعية الدموية، مما يخلق الشروط المسبقة لضعف تنظيم نغمة الأوعية الدموية، وزيادة ضغط الدم، وانتهاك تكيف القلب مع النشاط البدني. بالإضافة إلى ذلك فإن بعض الأطفال ذوي القامة الطويلة يعانون من مشاكل نفسية واجتماعية.
تجدر الإشارة إلى أن النمو المرتفع، وبشكل رئيسي، معدلات النمو السريع يمكن أن تكون علامة على أمراض الغدد الصماء المختلفة - مثل الورم الجسدي والبلوغ المبكر والتسمم الدرقي.
تعريف الطول
يتم إجراء تقييم موضوعي لنمو الطفل باستخدام درجة الانحراف المعياري (SDS، درجة الانحراف المعياري)، والتي توضح عدد انحرافات سيجما عن المعيار السكاني.
يتم تشخيص النمو عند الأطفال عندما يتجاوز طول الجسم أكثر من انحرافين معياريين (SD)، والعملقة - بأكثر من 3 انحراف معياري (SD) لجنس معين وعمر زمني معين.
بالإضافة إلى تقييم النمو، عند الأطفال ذوي القامة الطويلة، من الضروري حساب معدل النمو وتقييم وجود اللياقة البدنية غير المتناسبة والطول المتوقع النهائي. لحساب معدل النمو، من الضروري الحصول على بيانات عن قياسين موثقين للنمو خلال فترة لا تزيد عن 6 أشهر من أجل تسوية الأخطاء المحتملة في الحسابات. تجدر الإشارة إلى أن تقييم معدلات النمو هو الأكثر إفادة عند الأطفال قبل سن البلوغ.
لتقييم مدى تناسب الجسم، يتم قياس الارتفاع أثناء الجلوس ويتم تقييم نسبة الجزء العلوي من الجسم إلى الجزء السفلي.
يمكن حساب الارتفاع النهائي المتوقع للطفل بناءً على طول والديه وقيم العمر العظمي المحقق.
التشخيص التفريقي للقامة الطويلة
في معظم الحالات، لا يعد النمو المرتفع عند الأطفال أمرًا مرضيًا، ولكن المهمة الرئيسية لطبيب الغدد الصماء لدى الأطفال هي إجراء التشخيص التفريقي للأمراض الأخرى، والتي تتمثل أعراضها في ارتفاع معدلات النمو.
عند جمع سوابق المريض، ينبغي إيلاء الاهتمام للطول والوزن عند الولادة. وهكذا، مع متلازمات مارفان، وبيكويث-فيدمان، وسيمبسون-جولابي-بيميل، وبانايان-رايلي-روفالكابا والطول الدستوري، لوحظت قيم عالية لطول الجسم عند الولادة. تتميز معظم أشكال متلازمات القامة الطويلة (متلازمات سوتوس ويفر، متلازمة كلاينفلتر، متلازمة X الهشة، بيلة هوموسيستينية، وما إلى ذلك) بتأخر النمو النفسي العصبي أو مشاكل سلوكية. من المهم معرفة ما إذا كان المريض يعاني من أي أمراض مصاحبة. على سبيل المثال، تعتبر الاعتلالات الواقعية من سمات متلازمة مارفان والبيلة الهوموسيستينية. تعتبر أمراض الجهاز القلبي الوعائي أكثر شيوعًا في متلازمة مارفان، ومتلازمة لويس ديوتز، والبيلة الهوموسيستينية. انخفاض ضغط الدم لدى الأطفال حديثي الولادة شائع في متلازمة سوتوس، وعيوب جدار البطن الأمامي شائعة في متلازمة بيكويث-فيدمان. يعد توضيح ارتفاع الوالدين أمرًا مهمًا ليس فقط عند حساب الارتفاع المستهدف، ولكن أيضًا للتشخيص التفريقي للأشكال المتلازمية ذات القامة الطويلة. عند تحليل مؤشرات القياسات البشرية، من المهم تقييم مدى تناسب اللياقة البدنية. يعد تكوين اختلالات الجسم أمرًا نموذجيًا بالنسبة لمتلازمات مارفان وكلينفيلتر ولويس ديتز والبيلة الهوموسيستينية والعنكبوتية الانقباضية الخلقية (متلازمة بيلز) ، في حين أن المرضى الذين يعانون من كروموسوم X "الهش" ومتلازمات سوتوس ويفر وبيكويث فيدمان لديهم نسبة طبيعية من الأقسام العلوية والسفلية. تعتبر محيطات الرأس الكبيرة من سمات متلازمات سوتوس، ويفر، وبانايان-رايلي-روفالكابا.
يعد تقييم التطور الجنسي هو الجانب الأكثر أهمية في تحديد سبب طول القامة عند الأطفال. يشير النمو المتسارع مع الأرش عند الفتيات تحت سن 8 سنوات إلى التطور الجنسي المبكر. تشير معدلات النمو المرتفعة عند الأولاد، بالإضافة إلى زيادة حجم الخصية، إلى وجود نمو جنسي سابق لأوانه. ومع ذلك، لا ينبغي لنا أن ننسى أن الخصية الكبيرة مع القامة الطويلة هي مظهر سريري لمتلازمة X الهشة. تشمل أمراض الغدد الصماء الأخرى المرتبطة بمعدلات النمو المرتفعة فرط نشاط الغدة الدرقية وعملقة الغدة النخامية. يتم عرض تشخيص هذه الأمراض وغيرها المرتبطة بالقامة الطويلة في الجدول 1. بالإضافة إلى ذلك، تعد معدلات النمو المتسارع نموذجية للأطفال الذين يعانون من السمنة الخارجية الدستورية.
الجدول 1. التشخيص التفريقي للمكانة الطويلة عند الأطفال
|
مرض |
الاختبارات التشخيصية |
|
متلازمة مارفان |
الفحص من قبل طبيب عيون وطبيب قلب وعالم وراثة باستخدام معايير غنت |
|
متلازمة كلاينفلتر |
التنميط النووي |
|
متلازمة X الهشة |
البحوث الوراثية (جين نشرة الهجرة القسرية1) |
|
بيلة هوموسيستينية |
مستوى الهوموسيستين في الدم |
|
متلازمة سوتوس |
استخدام معايير خاصة، عمر العظام، الاستشارة الوراثية، اختبار الجينات NSD1 |
|
متلازمات أخرى |
الاستشارة الوراثية، والاختبارات الجينية المحددة |
|
التطور الجنسي المبكر |
LH، FSH، استراديول (للبنات)، التستوستيرون (للأولاد)، عمر العظام |
|
الكظر المبكر |
الأندروجينات الكظرية، التستوستيرون، استراديول، AFP، B-hCG، عمر العظام |
|
عملاقة الغدة النخامية |
GR، IGF-1، IGFBP3، اختبار قمع الجلوكوز (OGTT) |
|
فرط نشاط الغدة الدرقية |
TSH، شارع T4، شارع T3. |
|
نقص الجلايكورتيكويد العائلي |
ACTH، الكورتيزول |
|
نقص هرمون الاستروجين |
LH، FSH، استراديول، عمر العظام |
|
القامة الدستورية |
عمر العظام |
خيارات العلاج للمكانة الطويلة عند الأطفال
الغالبية العظمى من الأطفال ذوي القامة الطويلة لا يحتاجون إلى علاج محدد (لإيقاف النمو)؛ يوصى بالمراقبة الديناميكية مع مراقبة معدلات النمو.
إن مسألة إمكانية علاج القامة الطويلة مثيرة للجدل، لأنه حتى الآن لا توجد بيانات مقنعة عن فعاليتها وسلامتها. وبالإضافة إلى ذلك، فإن الشروط والأحكام التي يمكن بموجبها إجراء هذا العلاج لم يتم تحديدها بدقة. وفقًا لأغلبية الخبراء، من الممكن إجراء علاج محدد للقامة الطويلة عندما يتجاوز الارتفاع المتوقع أكثر من 2.5 انحراف معياري من القيم السكانية. الطرق الرئيسية لعلاج القامة الطويلة عند الأطفال هي الجراحية والطبية.
العلاج الهرموني الأكثر استخدامًا هو استخدام جرعات عالية من المنشطات الجنسية، مما يعزز الإغلاق السريع لصفائح النمو. في دراسة أجراها هندريكس أ. وآخرون. تبين أن استخدام جرعة عالية من هرمون الاستروجين (100 ميكروغرام من إيثينيل استراديول) فعال في علاج القامة الطويلة لدى الفتيات طويل القامة. كانت درجة الانخفاض في الارتفاع المتوقع النهائي متغيرة للغاية وتعتمد بشدة على عمر العظام الذي بدأ فيه العلاج. كلما بدأ العلاج لاحقًا، قل الانخفاض في الارتفاع النهائي. بالإضافة إلى ذلك، كان العلاج بالإستروجين بجرعات عالية مصحوبًا بآثار جانبية مثل التثدي عند الأولاد والغثيان والصداع وارتفاع ضغط الدم داخل الجمجمة والميل إلى تجلط الدم لدى المرضى من كلا الجنسين. تشير الدراسات المستقبلية في السنوات الأخيرة إلى الآثار الجانبية المتأخرة للعلاج بالإستروجين في شكل انخفاض الخصوبة لدى النساء. إن استخدام جرعات أعلى من هرمون الاستروجين (200 ميكروغرام من إيثينيل استراديول) لا يوفر فائدة في تقليل الطول النهائي ويرتبط بفشل المبيض الأولي وزيادة خطر الإصابة بسرطان الثدي لدى النساء. يرتبط استخدام جرعات عالية جدًا من هرمون الاستروجين (250 إلى 1000 ملغ) بزيادة كبيرة في خطر الإصابة بسرطان الجلد لدى النساء.
بالنسبة للأولاد طوال القامة، تُستخدم مستحضرات التستوستيرون بجرعات عالية (500 مجم، في العضل كل أسبوعين) لتقليل الطول النهائي المتوقع، لكن فعاليتها أقل بكثير من العلاج بالإستروجين. تعتمد فعالية العلاج أيضًا على درجة نضج العظام التي بدأ العلاج فيها. تشمل الآثار الجانبية للعلاج بالأندروجين حب الشباب والسلوك العدواني والانتصاب المؤلم التلقائي. يتم أيضًا استخدام علاج مماثل بنجاح في المرضى الذين يعانون من متلازمة مارفان دون أمراض القلب والأوعية الدموية. لم يتم الحصول على بيانات مقنعة عن زيادة الإصابة بالسرطان (بما في ذلك سرطان البروستاتا) وانخفاض الخصوبة لدى الرجال بعد تناول جرعات عالية من الأندروجينات.
إن ارتفاع معدل حدوث الآثار الجانبية للعلاج بالستيرويد الجنسي يدفع إلى إجراء دراسات حول استخدام أدوية أخرى تمنع الإفراز المركزي لهرمون النمو (نظائر السوماتوستاتين أو أدوية مضادات الكولين) أو تمنع العمل المحيطي لهرمون النمو.
الإجراء الجراحي الأكثر شيوعًا الذي يهدف إلى تقليل الارتفاع النهائي هو تثبيت المشاش عن طريق الجلد الثنائي للمشاشات الفخذية البعيدة والمشاشات الظنبوبية والشظية القريبة. تعتمد نتيجة العلاج الجراحي أيضًا على وقت العلاج. ولتقليل الطول النهائي المتوقع بمقدار الثلث، يجب إجراء العلاج الجراحي عند عمر عظمي يصل إلى 12.5 عامًا عند الفتيات بارتفاع يصل إلى 170 سم و14 عامًا عند الأولاد بارتفاع يصل إلى 185 سم. عند إجراء العلاج الجراحي، هناك خطر تشكيل اختلالات في الجسم، لأن نتيجة العلاج هي توقف نمو الأطراف السفلية، ولكن ليس العمود الفقري. مضاعفات ما بعد العملية الجراحية (الأعران، التشوهات الزاوية في الأطراف السفلية) تكون غائبة عمليا عندما يتم إجراء العملية من قبل جراح ذي خبرة. هناك تجربة إيجابية مع استخدام المشاش في المرضى الذين يعانون من متلازمة مارفان.
خاتمة
القامة الطويلة عند الأطفال ليست في أغلب الأحيان حالة مرضية تتطلب علاجًا محددًا. تتمثل المهمة الرئيسية لطبيب الغدد الصماء لدى الأطفال في تحديد سبب طول القامة، وهو ما يحدد المزيد من التكتيكات.
العلاجات الرئيسية للمكانة الطويلة هي العلاج الهرموني مع المنشطات الجنسية والعلاجات الجراحية. العلاج بجرعة عالية من هرمون الاستروجين لدى الفتيات ذوات القامة الطويلة له فعالية منخفضة ويرتبط بآثار جانبية خطيرة، مما يحد بشكل كبير من استخدامه في الممارسة السريرية اليومية. تعتبر الطرق الجراحية أكثر تفضيلاً لعلاج طويل القامة، ولكن لم يتم بعد دراسة سلامة وفعالية طريقة العلاج هذه بشكل كافٍ.
في تحضير هذه المادة استخدمنا:
Hannema S. E., Sävendahl L. تقييم وإدارة الطبيعة الشاهقة. هورم ريس بيدياتر 201؛85:347-352.
430 الفصل 13 أمراض الغدد الصم العصبية
هرمون النمو وIGF-1، مستوى هرمون النمو على خلفية OGTT
أكثر من 2 نانوجرام/مل، أو علامات التصوير بالرنين المغناطيسي أو الأشعة المقطعية للورم الغدي النخامي.
يتميز الأطفال حديثو الولادة المصابون بمتلازمة سوتوس (العملقة الدماغية) بزيادة في وزن الجسم وطوله، وجبهة محدبة، وتضخم الرأس، وضخامة الرأس، وارتفاع الحنك القوطي، وفرط التباعد مع ترتيب مضاد للمنغوليين في الشقوق الجفنية، والبروغناثيا. يعاني هؤلاء الأطفال من التخلف العقلي وضعف التنسيق الحركي. الجلد و
الأنسجة الرخوة سميكة إلى حد ما. مع التقدم في السن، يزداد نمو الجسم
يحدث، ولكن البلوغ عادة ما يحدث في وقت سابق، ونتيجة لذلك تغلق المشاشات في العظام الأنبوبية. هذا هو السبب في أن معظم المرضى الذين يعانون من المتلازمة المشتركة
توسا في مرحلة البلوغ يكون النمو الطبيعي.
تنعكس الصورة السريرية لمتلازمة بيكويث فيدمان بشكل جيد من خلال اسم آخر مقبول للمتلازمة - متلازمة EMG (قيلة سرية، ضخامة اللسان، عملاقة - من اللغة الإنجليزية. Exomphalos، Macroglossia، العملقة). في الواقع، يعاني الوليد المصاب بمتلازمة بيكويث فيدمان من أحجام كبيرة، وقيلة سرية، وتضخم اللسان، وتضخم الطبقة النخاعية في الكلى، وفرط التنسج
فجوة خلايا جزيرة البنكرياس، والتي غالبا ما تصبح السبب
تطور نقص السكر في الدم.
المظهر النموذجي للمرضى الذين يعانون من متلازمة مارفان أو متلازمات شبيهة بمارفان (بيلة هوموسيستينية، متلازمة MEN 2b،
العنكبوتية الانكماشية الخلقية) ، في المقام الأول
بسبب انخفاض الجزء العلوي من الجسم (ارتفاع الجلوس) بالنسبة للجزء السفلي، بما يتجاوز طول الجسم والعنكبوتية.
للحصول على الصورة السريرية لمتلازمة MEN 2b، راجع قسم "Syn-
"قلة من أورام الغدد الصماء المتعددة من النوع 2."
للحصول على الصورة السريرية لمتلازمة كلاينفلتر، انظر
"متلازمة البلوغ المتأخر."
بالإضافة إلى ذلك، يرتبط النمو المرتفع بزيادة خطر التطور
تطور بعض الأمراض والاضطرابات: الجنف مجهول السبب واضطرابات الوضع الأخرى، دوالي الخصية، استرواح الصدر العفوي، الاكتئاب، هشاشة العظام، الخ.
المسببات
القامة الدستورية.
أمراض الغدد الصماء.
GDM أو مرض السكري اللا تعويضي لدى الأم أثناء الحمل.
ورم سوماتوتروبيني في الغدة النخامية (عملقة الغدة النخامية) وأسباب أخرى لفرط إفراز هرمون النمو.
متلازمة PPS.
متلازمة الانسمام الدرقي.
خارجية دستوريةبدانة.
متلازمة قصور الغدد التناسلية.
المتلازمات الوراثية بما في ذلك الورم الجسدي
الغدة النخامية
متلازمة مين 1 (متلازمة فيرمر).
متلازمة ماكون أولبرايت.
متلازمة كارني.
ضخامة النهايات العائلية.
المتلازمات الوراثية التي تتميز بمكانة طويلة.
متلازمة سوتوس (العملقة الدماغية).
متلازمة ويفر سميث.
متلازمة بيكويث فيدمان.
متلازمة سيمبسون – جولابي – بيميل.
متلازمة مارفان.
متلازمة الرجال 2 ب.
بيلة هوموسيستينية.
العنكبوتية الانكماشية الخلقية (متلازمة بيلز).
متلازمة X الهشة (متلازمة مارتن بيل).
متلازمات مقاومة ACTH.
نقص الجلايكورتيكويد العائليالنوع الأول.
متلازمة "3A" (متلازمة ألجروف).
متلازمات مقاومة الاندروجين / الاستروجين ونقص الهرمونات (متلازمة تأنيث الخصية، وما إلى ذلك).
الحثل الشحمي المعمم الخلقي (lipoatro-
مرض السكري الجسدي، متلازمة سيب لورانس، متلازمة بيراردينيلي سيب).
الورم العصبي الليفي من النوع الأول (مرض ريكلينغهاوزن).
متلازمة كلاينفلتر.
متلازمة XYY (متلازمة فريق كرة السلة في السجن)
التشخيص
إذا كان لدى المولود الجديد طول زائد في جسمه، فيجب أن تكون الخطوة الأولى هي:
الخطوة الأولى هي البحث عن مرض السكري لدى الأم. إذا كانت الأم غير مصابة بالسكري
من الضروري استبعاد الأمراض الخلقية أولاً
بما في ذلك متلازمات سوتوس وبيكويث-فيدمان.
خوارزمية التشخيص
في مرحلة الطفولة، يتم تشخيص القامة الطويلة وفقًا للخوارزمية الموضحة في الشكل. 13-1.
تأكد من معرفة: ارتفاع الوالدين والأشقاء؛ الطول والوزن عند الولادة؛ معدل النمو طوال الحياة.
الفحص البدني
أثناء الفحص البدني، يتم إجراء ما يلي:
قياس الطول ومعدل النمو ووزن الجسم.
تقييم وجود وصمة العار الناتجة عن التفكك (السمات الخلقيةمنطقة الوجه والفكين، والأذنين، وما إلى ذلك)؛
التنمية الفكرية؛
432 الفصل 13 أمراض الغدد الصم العصبية
أرز. 13-1. خوارزمية التشخيص.
التطور الجنسي
نسبة طول الجزء العلوي من الجسم إلى طول الجزء السفلي؛
نسبة طول الذراع إلى طول الجسم.
الدراسات المخبرية والآلية
يشمل:
تحديد النمط النووي.
فحص الدم الهرموني(IGF-1، TSH)؛
تقييم عمر العظام.
حساب النمو النهائي المقدر.
اختبار البول للهوموسيستين.
الوراثية الجزيئيةبحث.
في الجدول 13-6 يشير إلى المبادئ الأساسية للتشخيص
الأمراض الرئيسية المسببة لطول القامة.
الجدول 13-6. مبادئ تشخيص الأمراض الرئيسية المسببة للقامة الطويلة
بحث، |
||
مرض |
السماح |
نتيجة |
تأكيد التشخيص |
||
ورم موجه جسدي |
مستويات هرمون النمو |
مستويات هرمون النمو في الخلفية |
الغدة النخامية |
على خلفية OGTT، التصوير بالرنين المغناطيسي / التصوير المقطعي |
OGTT > 2 نانوجرام/مل، التصوير بالرنين المغناطيسي أو التصوير المقطعي المحوسب- |
الغدة النخامية |
علامات ورم الغدة النخامية |
|
تحديد النمط النووي |
تحديد إضافية |
|
كلاينفلتر |
الكروموسومات X عند الرجال |
|
متلازمة XYY |
تحديد النمط النووي |
تحديد إضافية |
كروموسومات Y عند الرجال |
||
بيلة هوموسيستينية |
اختبار البول للهوموسيستين |
المستوى الأعلى |
الهوموسيستين في البول |
يعتمد تشخيص الأمراض الأخرى التي تسبب طول القامة على الصورة السريرية المميزة ونتائج الدراسات الوراثية الجزيئية.
علاج القامة الدستورية الطويلة
ويعتبر العلاج مناسبا إذا كانت النافذة المتوقعة
النمو النهائي لأكثر من ثلاثة انحرافات معيارية (للأفراد
العرق القوقازي أكثر من 195 سم للأولاد و 180 سم للفتيات). مؤشرات وصف العلاج الدوائي هي:
الجنف مجهول السبب والمشاكل النفسية والاجتماعية المرتبطة بالنمو المرتفع.
لعلاج القامة الطويلة، يمكن إبطاء النمو قبل البلوغ.
النمو بحيث يبدأ البلوغ في الوقت المناسب عند ارتفاع أقل، أو يقلل من نمو ما قبل البلوغ، مما يسرع البداية
بلوغ.
يهدف العلاج بالهرمونات الجنسية إلى تسريع بداية البلوغ. إذا بدأ العلاج مبكرًا، يزداد احتمال انخفاض الطول النهائي. أنسب وقت لوصف العلاج هو مرحلة ما قبل البلوغ
434 الفصل 13 أمراض الغدد الصم العصبية
الفترة (العمر العظمي للفتيات - 10 وللأولاد - 12.5 سنة).
العلاج بعد بداية البلوغ لم يعد فعالا.
تيفنا. يمكن أن يؤدي إعطاء الهرمونات الجنسية في عمر العظام الأكبر من 14 عامًا إلى زيادة الطول النهائي.
علاج القامة الطويلة البنيوية مع نظائر السوماتوستاتين ومضادات مستقبلات هرمون النمو لا يستخدم بعد في الممارسة السريرية واسعة النطاق.
علاج البنات
يتم وصف الفتيات ذوات القامة الدستورية
إيثينيل استراديول عن طريق الفم 0.15-0.3 ملغ مرة واحدة في اليوم. إذا لزم الأمر، يمكن زيادة جرعة إيثينيل استراديول إلى 0.5 ملغ / يوم إذا كان المريض يتحمل الدواء جيدًا. تطبيق يخدع
كما أن هرمون الاستروجين المقتطف (7.5-10 ملغ/يوم) فعال أيضًا
فعال. في حالة حدوث نزيف الرحم، تتم الإشارة إلى الاستخدام الدوري للهرمونات الجنسية؛ يضاف الديدروجستيرون عن طريق الفم 10 ملغ مرتين في اليوم إلى هرمون الاستروجين من اليوم السادس عشر إلى اليوم الخامس والعشرين من الدورة الشهرية أو النوريثيستيرون عن طريق الفم 5 ملغ مرة واحدة يوميًا من اليوم السادس عشر إلى اليوم الخامس والعشرين. اليوم الرابع من الدورة الشهرية. يجب أن يستمر العلاج حتى تغلق المشاشات، وإلا
قد يستمر النمو بعد انسحاب هرمون الاستروجين.
إذا كان تحمل إيثينيل استراديول ضعيفًا، يمكن استبدال استراديول فاليرات (تتم زيادة الجرعة تدريجيًا من 2 إلى 6 ملغ / يوم).
علاج الأولاد
في الممارسة العملية، فإن الحاجة إلى علاج الأولاد بأدوية التستوستيرون نادرة للغاية.
التستوستيرون (خليط من الاسترات) في العضل 250 ملغ مرة واحدة في اليوم
أسبوع أو 500 ملغ مرة واحدة كل أسبوعين لمدة 6-12 شهرًا.
علاج طول القامة في أمراض أخرى
تستخدم الهرمونات الجنسية للحد من نمو الخلايا المتزامنة.
قرع مارفان.
بالنسبة لمتلازمات بيكويث فيدمان وسوتو، فهي أعراض
العلاج ماتيتش.
علاج ورم الغدد التناسلية الجسدية - انظر "ضخامة النهايات وعملقة الغدة النخامية".
علاج متلازمة MEN 2b - راجع "متلازمة أورام الغدد الصماء المتعددة من النوع 2".
علاج متلازمة كلاينفلتر - انظر "متلازمة البلوغ المتأخر"، "متلازمة كلاينفلتر" (فرط الغدد التناسلية)
قصور الغدد التناسلية المدارية).
متلازمة المماطلة القصيرة
تصنيف
نقص هرمون النمو مجهول السبب.
نقص هرمون النمو مجهول السبب (الكلاسيكي
ضعف الإفراز العصبي (NSD).
نقص هرمون النمو من أصل عضوي: خلقي
الشكل المعطى والمكتسب.
أسباب أخرى لقصر القامة
قصر القامة مجهول السبب: عائلي، غير عائلي.
المتلازمات المحددة سريريًا مع انحراف الكروموسومات: تيرنر، داون، خلل تكوين الغدد التناسلية.
المتلازمات المحددة سريريًا دون انحراف الكروموسومات:سيلفر راسل، نونان، ريكلينغهاوزن، دي لانج،
ويليامز، بلوم، برادر ويلي، روبنشتاين.
تأخر النمو داخل الرحم مع قصر القامة بعد الولادة، دون وصمات عار.
تقييد النمو داخل الرحم مع قصر القامة بعد الولادة
الخسارة، مع الوصمات: بسبب التهابات الفترة المحيطة بالولادة،
تعاطي الأم للمخدرات أو التدخين أو تعاطي الكحول، وما إلى ذلك.
خلل التنسج الهيكلي: الودانة، نقص التنسج الغضروفي، تكون العظم الناقص، إلخ.
أمراض استقلاب العظام: داء عديد السكاريد المخاطي، داء الشحميات المخاطية، إلخ.
الأمراض الاستقلابية: مرض تخزين الجليكوجين،
مرض تخزين الدهون، بيلة الفينيل كيتون، الخ.
قصر القامة علاجي المنشأ.
التشخيص
انظر "قياس وتقييم النمو".
يتم تحديد نقص النمو باستخدام جداول التباين المئوية.
النمل من الارتفاع الطبيعي. تحليل منحنى نمو الطفل مع
مع مراعاة حدود طوله النهائي، محسوبة على أساس متوسط طول والديه:
الارتفاع النهائي المتوقع للصبي = [(طول الأب + طول الأم + 13 سم)/2] ± 10 سم الارتفاع النهائي المتوقع للفتاة =
[(طول الأب + طول الأم – 13 سم)/2] ± 10 سم.
إذا كان الطول النهائي المستقر للطفل يعتمد على بيانات الميلاد
المائة وقت الفحص مع مراعاة عمر العظام تكون أقل
حدود فترة النمو النهائية المحسوبة، ثم يلي ذلك
الحديث عن النمو المنخفض بشكل مرضي.
من المهم أولاً تقييم مدى تناسب الهيكل العظمي
لاستبعاد أشكال مختلفة من خلل التنسج الهيكلي كأسباب لقصر القامة. على وجه الخصوص، يُنصح بحساب معامل "الجزء العلوي/السفلي" وامتداد الذراع.
تعد درجة تعظم مناطق النمو المشاشية معيارًا مهمًا في تشخيص التقزم والتشخيص للنمو النهائي.
في حالة نقص النمو الأولي، يكون التأخر في نضوج العظام إما غائبًا أو خفيفًا (خلل التنسج الهيكلي، الأشكال المتلازمية من التقزم، تأخر النمو داخل الرحم،
436 الفصل 13 أمراض الغدد الصم العصبية
قصر القامة الوراثي). لنقص النمو الثانوي،
وخاصة بالنسبة للقزامة النخامية، كبيرة
تأخر عمر العظام عن العمر الزمني (> سنتين).
فحص الأشعة السينية للجمجمة تم إجراؤها من أجل تصور شكل وحجم السرج التركي وحالة عظام الجمجمة. في حالة التقزم النخامي، غالبًا ما يكون السرج التركي صغير الحجم. لوحظت تغيرات مميزة في السرج التركي في الورم القحفي البلعومي: ترقق ومسامية الغشاء المخاطي.
nok، اتساع المدخل، فوق أو بؤر داخل النجوم عادة
رسالة. مع زيادة الضغط داخل الجمجمة، تظهر زيادة في الانطباعات الرقمية وتباعد خيوط الجمجمة.
في التصوير المقطعي والتصوير بالرنين المغناطيسي للدماغفي المرضى الذين يعانون من مجهول السبب
يتم الكشف عن قصور الغدة النخامية من خلال المورفولوجية والهيكلية
التغيرات السياحية: نقص تنسج الغدة النخامية، تمزق أو ترقق ساق الغدة النخامية، انتباذ النخامية العصبية، متلازمة السرج الفارغة. يُستطب التصوير المقطعي المحوسب والتصوير بالرنين المغناطيسي للدماغ لأي اشتباه في وجود أمراض داخل الجمجمة (عملية احتلال الفضاء).
تأخر النمو البنيوي والبلوغ (CGRP) وقصر القامة العائلي (أو قصر القامة الطبيعي المتغير)
النمو) يتناسب مع مفهوم خيارات النمو الطبيعي.
الأطفال من آباء ذوي قصر القامة، كقاعدة عامة، يكونون قصيري القامة بنفس القدر مثل والديهم، وذلك بسبب إمكانات النمو المبرمجة وراثيا. الأطفال من الآباء الذين لديهم تاريخ
تأخر النمو الشديد والبلوغ، مع وجود درجة عالية من الاحتمال
سوف يرث هذا النمط التنموي.
يتمتع الأطفال المصابون بـ CGRP بطول ووزن طبيعيين عند الولادة، وينموون بشكل طبيعي حتى عمر 1-2 سنة، ثم ينخفض معدل النمو.
ومنحنى النمو أقل بقليل من المئين الثالث
والموازي لها. عمر العظام يتوافق عادة مع
عمر النمو ومعدل النمو - 5 سم على الأقل في السنة. في عينات ل
يكشف التحفيز عن إطلاق كبير لهرمون النمو (> 10 نانوجرام/مل)،
ولكن يقل الإفراز اليومي المتكامل لهرمون النمو في الدم. بو-
بيرتات طبيعي، ولكنه يتأخر بسبب تأخر عمر العظام ويحدث عندما يتم الوصول إلى نضج العظام عند الأولاد في عمر 11.5-12 سنة، وفي الفتيات في عمر 10.5-11 سنة. يتم تغيير توقيت تحقيق النمو النهائي مع مرور الوقت؛ وعادة ما يكون النمو النهائي
طبيعي وبدون علاج هرموني.
هناك أدلة على العلاج الفعال بالهرمونات المؤتلفة
احتكار النمو البشري للأطفال الذين يعانون من CGRP، وتأخر النمو داخل الرحم، وقصر القامة العائلي، ومتلازمات تيرنر، راسل سيلفر، برادر ويلي، فقر الدم فانكوني، مرض إتسينكو كوشينغ، داء الجليكوجين، مع حالة بعد التشعيع لسرطان الدم وأورام المخ، بعد الكلى زرع ، مع الفشل الكلوي المزمن ،
خلل التنسج الهيكلي. وفي الوقت نفسه، لا توجد بيانات موثوقة عن النمو النهائي العالي للأطفال الذين يتلقون CGRP
يعالجون بهرمون النمو مقارنة بمن لم يتلقوه بالرغم من ذلك
أنه على مدى فترة زمنية معينة (2-3 سنوات) يتم العلاج بـ GH
يسرع بشكل كبير معدل النمو.
في الأولاد الذين يعانون من CCRP الذين تزيد أعمارهم عن 12 عامًا مع تأخر في عمر العظام لمدة عامين على الأقل من العمر الزمني، من الممكن العلاج قصير الأمد بدورات قصيرة من جرعات صغيرة من الستيرويدات الابتنائية (nerobolil♠، retabolil♠). حيث
من الضروري إجراء مراقبة صارمة لنمو الطفل (مرة واحدة على الأقل كل يوم).
6 اشهر). إذا تقدم نضوج العظام بسرعة، يتم وقف العلاج.
قزامة الغدة النخامية (قصور جسدي)
إن قياس واحد لهرمون النمو في الدم لتشخيص القصور الجسدي ليس له أي قيمة تشخيصية بسبب الطبيعة النبضية لإفراز هرمون النمو وإمكانية الحصول على قيم قاعدية منخفضة للغاية (صفر)
GH حتى في الأطفال الأصحاء. في هذا الصدد، عفوية
إفراز هرمون النمو في الدم، وتحديد ذروة إطلاق هرمون النمو على خلفية التحفيز، وفحص IGFs والبروتينات المرتبطة بها في الدم.
تعتمد الاختبارات الاستفزازية على القدرة على التمييز
تحفز الأدوية الدوائية إفراز وإطلاق هرمون النمو عن طريق الجسديات. في الممارسة السريرية، الاختبارات الأكثر استخدامًا هي الأنسولين والكلونيدين والهرمون المطلق للـ GH والأرجينين والليفودوبا وبروميد البيريدوستيجمين (الجدول 13-7).
يتم تشخيص القصور الجسدي الكلي عندما يكون إطلاق ذروة هرمون النمو على خلفية التحفيز أقل من 7 نانوجرام/مل، جزئيًا
نقص كبير - في ذروة إطلاق هرمون النمو من 7 إلى 10 نانوغرام / مل.
شرط ضروري لتنفيذ تحفيز هرمون النمو
عينات - حالة الغدة الدرقية من الغدة الدرقية. في حالة قصور الغدة الدرقية، فمن الضروري
نحن نأخذ دورة علاجية أولية بأدوية الغدة الدرقية لمدة 3-4 أسابيع. بالإضافة إلى ذلك، فإن الأطفال الذين يعانون من السمنة المفرطة لديهم استجابة منخفضة للتحفيز.
للتقييم المتزامن للعديد من وظائف الغدة النخامية
ومن الملائم إجراء اختبارات مشتركة مع مختلف الهدايا
الهرمونات المطلقة للمهاد [الأنسولين + الهرمون المطلق للثيروتروبين
(TRH) + اللوليبرين (اختبار LHRH)، الهرمون المطلق لـ GH + TRH +
اختبار LHRH، الهرمون المطلق للـ GH + الكورتيكوليبرين (CRH) +
اختبار LHRH + TRH]. على سبيل المثال، عند الاختبار باستخدام الهرمون المطلق لـ GH + الثايروليبيرين + اللوليبرين، يتم حقن الهرمون المطلق للـ GH (1 ميكروجرام/كجم)، والثيروليبيرين (7 ميكروجرام/كجم، بحد أقصى 400 ميكروجرام)، واللوليبيرين (100 ميكروجرام) بشكل تسلسلي في القنية الوريدية . يشير وجود مستويات قاعدية منخفضة من TSH وT4 الحر مع غياب أو استجابة طويلة من TSH إلى TRH إلى قصور الغدة الدرقية الثانوي المصاحب. غياب استجابة إطلاق موجهة الغدد التناسلية لهرمون LHRH (زيادة أكثر من ضعف المستوى الأساسي) بالاشتراك مع انخفاض
تشير المستويات القاعدية للهرمونات الجنسية إلى مستوى ثانوي
قصور الغدد التناسلية.
الفصل 13 أمراض الغدد الصم العصبية |
||||||
الجدول 13-7. اختبارات تحفيز السوماتوتروبين |
||||||
العقار |
آلية |
الجرعة، الطريقة |
آثار جانبية |
|||
أجراءات |
مقدمة |
|||||
عينات من الدم |
||||||
مقدمات |
||||||
التنشيط |
0.1 وحدة/كجم، رابعا |
نقص سكر الدم |
||||
تحت المهاد |
||||||
الخلايا العصبية للتزلج, |
||||||
تنشيط |
||||||
إفراز هرمون النمو |
||||||
إطلاق- |
||||||
الكلونيدين |
الناقل العصبي- |
0.15 ملجم/م2، |
شرياني |
|||
ثالثا، الأدرينالية |
انخفاض ضغط الدم, |
|||||
ناهض منطقي |
النعاس |
|||||
تحت المهاد |
1 ميكروجرام/كجم، وريديًا |
|||||
إطلاق- |
إطلاق السماء- |
|||||
ليفودوبا |
الناقل العصبي- |
|||||
ثالثا، الدوبامين- |
(كتلة الجسم |
|||||
قبل سحرية- |
صداع |
|||||
نيست. يحفز- |
||||||
لا الافراج |
(كتلة الجسم |
|||||
إطلاق STG- |
||||||
(كتلة الجسم |
||||||
إل-أرجينين |
الأيض |
نقص سكر الدم، |
||||
هيدروكلورو- |
منبه, |
احمرار |
||||
حمض أميني. |
||||||
يحفز |
حل |
|||||
تحرير |
||||||
إطلاق STG- |
||||||
الرابع، التسريب |
||||||
خلال |
||||||
الجلوكاجون |
نسبي |
100 ميكروغرام/م2، |
||||
نقص سكر الدم |
أقصى |
القيء، في وقت متأخر |
||||
نقص سكر الدم |
||||||
الثوابت الأكثر أهمية تشخيصيا في تحديد
نقص هرمون النمو لدى الأطفال - IGF، وخاصة IGF-1 (السوماتوميدين C) وIGF-2.
لعلاج قزم الغدة النخامية، يتم استخدام مستحضرات هرمون النمو البشري المعدلة وراثيا كعلاج هورموني بديل. حاليًا، تمت الموافقة على مستحضرات السوماتوتروبين التالية للاستخدام في روسيا:
جينوتروبين ♠ (جينوتروبين ♠) ؛
نورديتروبين سيمبلكس♠ (نورديتروبين سيمبلكس ♠);
سايزن ♠ (سايزن ♠);
هوماتروب ♠ (هوماتروب ♠);
الرستن ♠ .
الجلد يوميًا من الساعة 20:00 إلى الساعة 22:00.
يجب أن يستمر العلاج حتى يتم إغلاق مناطق النمو أو حتى
تحقيق نمو مقبول اجتماعيا.
عندما يتم الجمع بين التقزم النخامي ونقص هرمون الغدة النخامية المتعددة، يكون ذلك مناسبًا
العلاج التعويضي بالهرمونات مع أدوية الغدة الدرقية (L- هرمون الغدة الدرقية)، الجلايكورتيكويدات
دامي (الهيدروكورتيزون)، والمنشطات الجنسية.
متلازمة شيرشيفسكي-تيرنر (خلل تكوين الغدد التناسلية)
قصر القامة هو المظهر السريري الأكثر شيوعا
ظاهرة متلازمة شيرشيفسكي-تيرنر. يجب الاشتباه في هذه المتلازمة في المقام الأول عند الفتيات اللاتي يعانين من تأخر النمو غير المبرر. مع الأخذ في الاعتبار الأشكال الفسيفسائية للمتلازمة
شيرشيفسكي-تيرنر أماه (45 ХО/46 ХХ، 45 ХО/46 Хi(Хq)، 46
Xi(Xq)، 45 XO/46 X، rX، وما إلى ذلك) مع مجموعة بسيطة من الأعراض السريرية النموذجية أو حتى غيابها، يُطلب من جميع الفتيات المصابات بتأخر النمو (الطول SDS ≥2 SDS) إجراء دراسة النمط النووي في المرحلة الأولى من البحث التشخيصي.
يتمثل نمط النمو لدى الفتيات المصابات بمتلازمة شيرشيفسكي-تيرنر في تأخر النمو المعتدل داخل الرحم.
(يبلغ متوسط طول ووزن الجسم عند الأطفال حديثي الولادة حوالي 1 SD
أقل من القيم الطبيعية ويبلغ متوسطها 48.3 سم و2800 جم على التوالي)، بمعدل طبيعي نسبياً
النمو من الولادة إلى 3 سنوات من الحياة، والانخفاض التدريجي
متوسط الطول لمتلازمة شيريشيفسكي-تيرنر هو
في المتوسط 142.0-146.8 سم.
بالإضافة إلى تأخر النمو، تشمل الأعراض السريرية
الوذمة اللمفاوية في اليدين والقدمين أثناء فترة حديثي الولادة ،
رقبة قصيرة ذات طيات على شكل جناح ذات تعبيرات مختلفة
انخفاض نمو الشعر في الجزء الخلفي من الرقبة، تدلي الجفون (عادة
ثنائي)، micrognathia، epicanthus، الحنك القوطي، مشوه
تزاوج الأذنين، والصدر على شكل برميل مع حلمات متباعدة على نطاق واسع، والجنف، وانحراف أروح المرفق
المفاصل، تشوه الرئة، تقصير وسماكة الأصابع، الخطوط الجلدية النموذجية، خلل تنسج الأظافر، الشامات المصطبغة المتعددة.
يتم إجراء الاختبارات الاستفزازية لتحفيز هرمون النمو فقط في المرضى الذين يعانون من متلازمة شيرشيفسكي-تيرنر، الذين يعانون من متلازمة شيريشيفسكي-تيرنر
ينحرف منحنى النمو عن ذلك الخاص بهذه المتلازمة.
معظم المرضى لديهم استجابة طبيعية لهرمون النمو للتحفيز خلال مرحلة الطفولة. انخفاض مستويات هرمون النمو في
من المحتمل أن ترتبط فترة البلوغ بمستويات منخفضة من النشاط الجنسي